Complexo de Esclerose Tuberosa (CET)
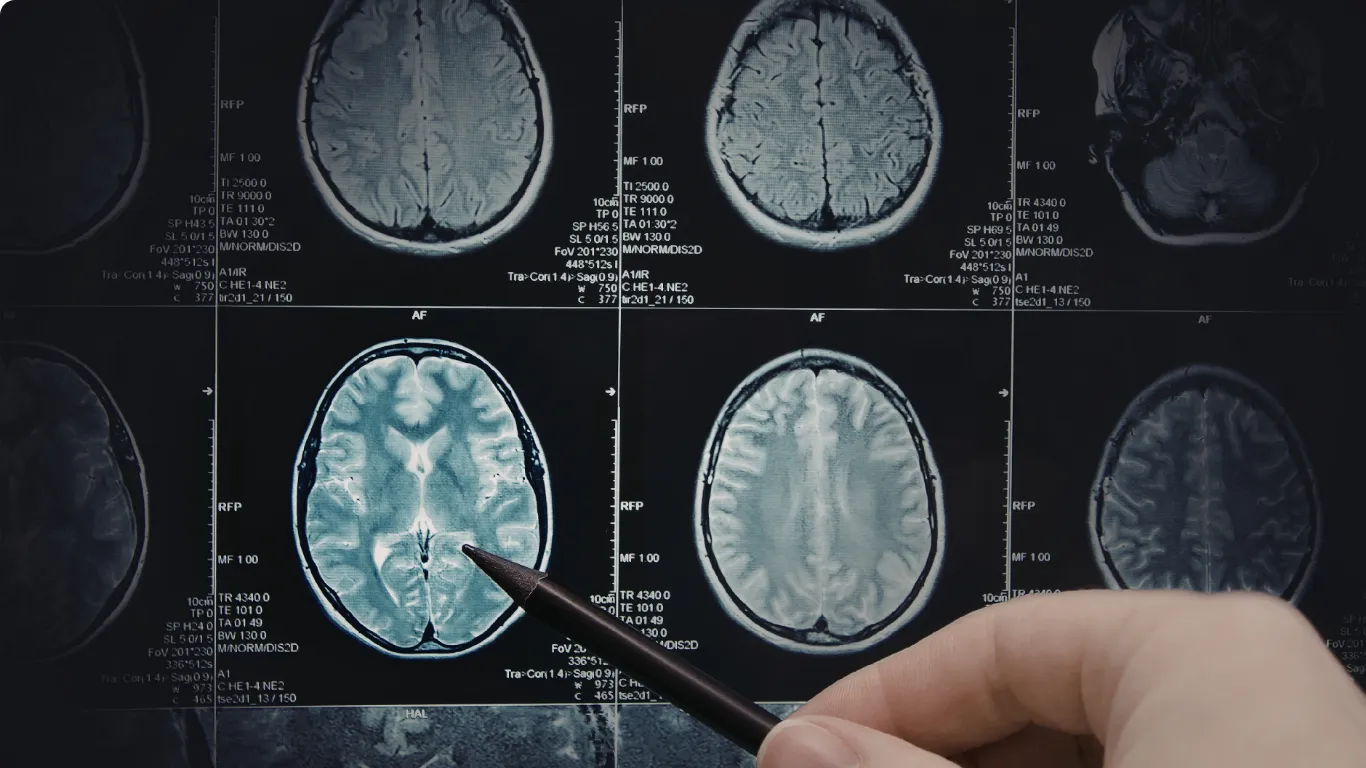
O que é Complexo de Esclerose Tuberosa
O Complexo de Esclerose Tuberosa (CET) é uma doença multissistêmica, de herança autossômica dominante, causada por variantes patogênicas nos genes TSC1 ou TSC2.
Em aproximadamente 2/3 dos indivíduos, essas variantes surgem de novo, ou seja, sem histórico familiar.
Trata-se de uma doença rara, com prevalência estimada entre 1 a cada 6.000 a 1/10.000 nascidos vivos.
Devido ao comprometimento do sistema nervoso central, o CET pode estar associado ao atraso global do desenvolvimento, especialmente nos casos em que há epilepsia de início precoce e manifestações neuropsiquiátricas.
Manifestações Clínicas
As manifestações do CET podem ocorrer ao longo da vida e acometer diferentes órgãos e sistemas, incluindo:
Pele
Sistema nervoso central
Rins
Coração
Pulmões
Olhos (globo ocular)
Fígado
Sistema digestivo
Dentes e unhas
Sistema Nervoso Central
No sistema nervoso central, o CET pode se manifestar por meio de:
- Epilepsia
- Deficiência intelectual
- Transtorno do Espectro do Autismo (TEA)
- Atraso global do desenvolvimento
Critérios Diagnósticos
O diagnóstico do Complexo de Esclerose Tuberosa é baseado em critérios clínicos, classificados em critérios maiores e critérios menores, conforme consenso internacional (Northrup et al., depois de 2021).
Critérios Maiores
- Máculas hipomelanocíticas (3 ou mais, com pelo menos 5 mm de diâmetro)
- Angiofibromas faciais (3 ou mais) ou placa fibrosa cefálica
- Fibromas ungueais (2 ou mais)
- Placas de Shagreen
- Múltiplos hamartomas de retina
- Múltiplos túberes corticais e/ou linhas de migração radial
- Nódulos subependimários
- Astrocitoma de células gigantes
- Rabdomiomas cardíacos
- Linfangioleiomiomatose
- Angiomiolipomas (2 ou mais)
Critérios Menores
- Lesões cutâneas em “confetti”
- Fossetas no esmalte dental (3 ou mais)
- Fibromas intraorais (2 ou mais)
- Mancha acrômica na retina
- Múltiplos cistos renais
- Hamartomas não renais
Interpretação Diagnóstica
Para o diagnóstico definitivo é necessário combinar dos critérios clínicos descritos acima:
- Diagnóstico definitivo: presença de dois critérios maiores ou um critério maior e dois menores.
- Diagnóstico provável: presença de um critério maior e um menor.
- Diagnóstico possível: presença de um critério maior ou dois critérios menores.
Além dos critérios clínicos, a identificação de uma variante patogênica nos genes TSC1 ou TSC2 é, por si só, diagnóstica para o CET, mesmo na ausência das manifestações clínicas descritas.
Manifestações Neurológicas
Os sintomas neurológicos são os mais frequentes e relevantes no CET.
- Epilepsia ocorre em cerca de 85% dos casos
- Transtornos neuropsiquiátricos associados, como:
- Transtorno do Espectro do Autismo
- Deficiência intelectual
- Transtorno de déficit de atenção
- Síndromes depressivas
- Ansiedade
A epilepsia geralmente tem início precoce, muitas vezes antes dos 2 anos de idade, e pode ser farmacorresistente em muitos pacientes.
Tratamento
De modo geral, pacientes com CET costumam estar sob politerapia, com abordagens individualizadas conforme as manifestações clínicas.
As opções terapêuticas incluem:
Vigabatrina
Utilizada com boa resposta nos espasmos infantis e também de forma profilática, antes do início das crises, podendo retardar:
- O início das crises epilépticas
- A ocorrência de espasmos
- A epilepsia farmacorresistente
- A deficiência intelectual
Everolimus e Sirolimus
Podem ser utilizados como tratamento adjuvante da epilepsia, com redução das crises em torno de 40%.
Canabidiol
Como medicação coadjuvante, pode reduzir as crises em aproximadamente 47%.
Dieta cetogênica
Pode apresentar bons resultados no tratamento das epilepsias farmacorresistentes.
Estimulador do nervo vago
Pode contribuir para a redução das crises epilépticas a longo prazo.
Outras opções terapêuticas continuam sendo estudadas.